
Consider a rare genetic condition that only a small number of people worldwide are afflicted with. The rare, life-altering Niemann-Pick Disease (NPD) affects the body’s capacity to metabolize and break down fats, resulting in serious complications. The illness gradually harms several organs, including the brain, liver, and spleen, and is frequently identified in infancy or early childhood.
An Intractable Genetic Mysteries
Niemann-Pick disease is fundamentally a lysosomal storage disorder, which means that the body is unable to appropriately eliminate and recycle lipids from cells. These fats build up over time, impairing organ function and causing a catastrophic deterioration in health. There is currently no cure, only supportive care to control symptoms and extend quality of life, while researchers continue to investigate possible treatments.
Types of Niemann-Pick Disease
| Type | Description | Age of Onset | Primary Symptoms |
|---|---|---|---|
| Type A | Most severe, primarily affects infants | Infancy | Enlarged liver and spleen, severe brain damage, early death |
| Type B | Milder form, primarily affecting pre-teens and adults | Childhood to adulthood | Liver and spleen enlargement, lung disease, minimal brain involvement |
| Type C | Progressive neurological disorder | Infancy to adulthood | Loss of motor function, speech issues, memory loss, cognitive decline |
Symptoms That Should Not Be Ignored
Early detection is critical. Symptoms of Niemann-Pick Disease can vary widely depending on the type and severity, but common warning signs include:
- Enlarged liver and spleen (often one of the first signs in infants)
- Loss of motor skills (difficulty walking, balancing, and coordinating movements)
- Frequent respiratory infections (chronic lung disease is common in Type B patients)
- Cognitive decline (affecting speech, memory, and ability to process information)
- Cherry-red spots in the eyes (a hallmark feature of Type A)
- Feeding difficulties and poor growth (leading to malnutrition)
What Causes Niemann-Pick Disease?
The condition is inherited through autosomal recessive genes, meaning both parents must carry a defective gene to pass it on to their child. The specific gene mutations differ by type:
- Types A & B result from mutations in the SMPD1 gene, which leads to a deficiency in the enzyme acid sphingomyelinase (ASM). Without ASM, fats accumulate in cells.
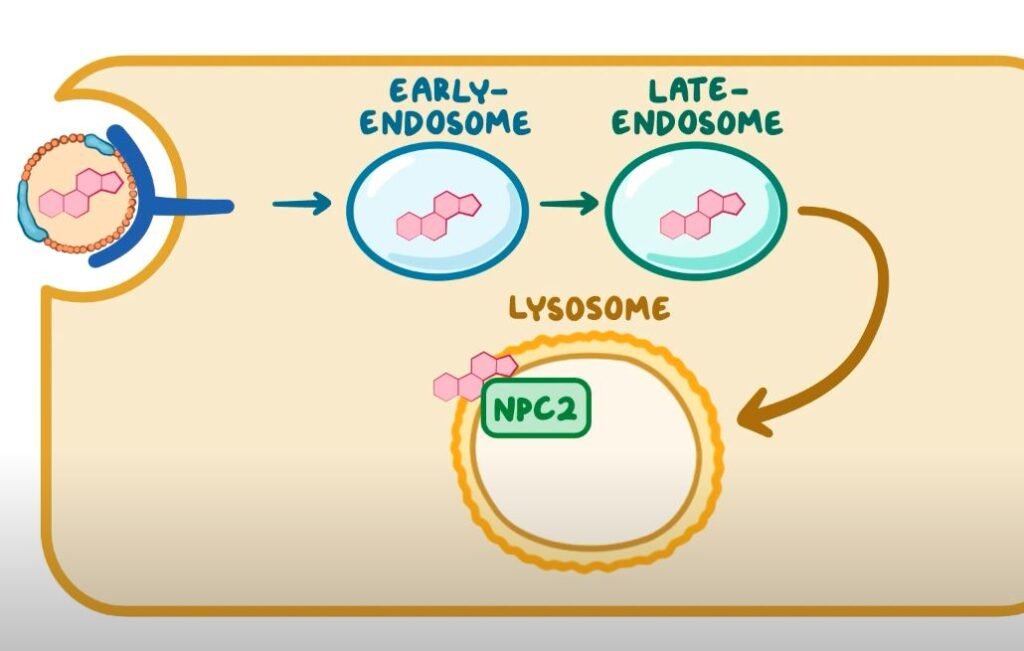
- Type C stems from mutations in the NPC1 or NPC2 genes, which disrupt cholesterol and lipid transport within cells.
Because it is a genetic disorder, families with a history of Niemann-Pick should seek genetic counseling to assess risks for future pregnancies.
The Challenge of Treatment: Managing, Not Curing
Currently, there is no cure for Niemann-Pick Disease, but several treatments aim to manage symptoms and slow progression:
1. Supportive Medical Care
- Statins to regulate lipid levels in Type B patients
- Physical therapy to maintain mobility and muscle function
- Speech therapy for patients experiencing cognitive decline
- Blood transfusions in cases of excessive bleeding due to low platelet counts
2. Enzyme Replacement and Gene Therapy Prospects
- Olipudase alfa (approved in Japan and the EU) is a promising enzyme replacement therapy for Type B
- Gene therapy research aims to correct the faulty genes responsible for the disease, though it is still in early-stage clinical trials
3. Experimental Drug Therapies
- Miglustat has been approved in Europe for Type C and shows promise in slowing neurological decline
- Cyclodextrin-based therapies are being studied for their potential to clear lipid accumulation in Type C
Living with Niemann-Pick Disease: The Emotional Cost
Niemann-Pick disease is a life-altering diagnosis for families. Providing care for a child with a degenerative disease can be financially, emotionally, and physically taxing. For impacted families, support groups, online forums, and specialized medical teams offer vital resources. Groups such as the National Niemann-Pick Disease Foundation (NNPDF) provide essential funding for patient support, advocacy, and research.
Niemann-Pick Research’s Prospects
Science is advancing. Advances in stem cell research, enzyme therapy, and genetic medicine raise the possibility that Niemann-Pick disease could one day be prevented or treated. In the upcoming years, Niemann-Pick treatment could undergo a significant transformation with sustained research and funding.
Questions and Answers (FAQs)
- Can someone die from Niemann-Pick disease?
The type determines this. While Type B patients can survive into adulthood, Type A patients typically die in their early years. Type C varies greatly in severity and advances over time.
- How is the diagnosis of Niemann-Pick disease made?
The diagnosis is confirmed by a combination of imaging tests, enzyme activity assays, and genetic testing.
- Is it possible to avoid Niemann-Pick disease?
Given that it is genetic, genetic counseling and carrier screening can assist parents in determining their risks prior to becoming parents.
- Does Niemann-Pick Disease have a treatment?
Not just yet. Clinical trials give hope for future advancements, but current treatments concentrate on managing symptoms and delaying progression.
- Which institutions fund Niemann-Pick studies?
Organizations such as the Niemann-Pick UK and the NNPDF (National Niemann-Pick Disease Foundation) contribute to the funding of research and offer support to impacted families.